Neurogenetics Department
ΥΠΗΡΕΣΙΕΣ, CMT:Γενετική Διάγνωση με τεχνολoγία NGS

Η νόσος Charcot-Marie-Tooth (CMT) ή κληρονομική κινητική και αισθητική νευροπάθεια ανήκει στην κατηγορία των πολυνευροπαθειών και προσβάλει τα περιφερικά νεύρα με αποτέλεσμα την παρεμπόδιση μετάδοσης πληροφορίων από τον εγκέφαλο και το νωτιαίο μυελό προς το υπόλοιπο σώμα, καθώς και πληροφορίες από το σώμα π.χ. αφή, προς στο νωτιαίο μυελό και τον εγκέφαλο. Αυτό έχει άμεσες συνέπειες και στα κινητικά νευρά των μυών προκαλώντας σταδιακά μυϊκή αδυναμία και ατροφία. Η νόσος CMT είναι από τις πιο κοινές κληρονομικές νευρομυϊκές διαταραχές, επηρεάζοντας περίπου 1 στα 2,500 άτομα. Τα κλινικά χαρακτηριστικά, η σοβαρότητα και η ηλικία έναρξης της νόσου ποικίλουν. Η εξέλιξη των συμπτωμάτων είναι σταδιακή. Ο τυπικός φαινότυπος περιλαμβάνει:
- Αδυναμία ή παράλυση των μυών των κάτω άκρων (ποδιών), που μπορεί να προκαλέσει πτώση ποδιού (δυσκολία στην ανύψωση του ποδιού)
- Μπορεί να εμφανιστεί αδυναμία και ατροφία των πάνω άκρων (χεριών), προκαλώντας δυσκολία στις λεπτές κινητικές δεξιότητες
- Καλπαστικό βάδισμα (όταν υπάρχει αμφοτερόπλευρο δίκροτο βάδισμα), με συχνό παραπάτημα ή πτώση
- Παραμορφώσεις του ποδιού, όπως χαρακτηριστικές ψηλές ποδικές καμάρες, σφυροδακτυλία, κλπ.
- Αισθητική απώλεια (ζέστης, κρύου και αφής) και μείωση ή απουσία τενόντιων αντανακλαστικών
- Προβλήματα ισορροπίας και μειωμένη αίσθηση δόνησης και θέσης
- Σκολίωση (παραμόρφωση) σπονδυλικής στήλης, μυϊκές κράμπες και πόνος των νεύρων
Σε μερικές περιπτώσεις τα άτομα εμφανίζουν απώλεια όρασης και/ή ακοής και δυσκολίες αναπνοής ενώ κάποιοι ασθενείς χρειάζονται ορθοπεδικά βοηθήματα.
Η CMT σύμφωνα με ηλεκτροφυσιολογικά και παθολογικά κριτήρια κατατάσσεται σε απομυελινωτικό, αξονικό και ενδιάμεσο τύπο. Στον απομυελινωτικό τύπο (CMT1) παρατηρείται καταστροφή της μυελίνης (προστατευτικό περίβλημα των νευρικών ινών) των περιφερικών νευρών ενώ στον αξονικό τύπο παρουσιάζεται κυρίως αξονική εκφύλιση. Επίσης βάσει του τρόπου κληρονόμησης η CMT μπορεί να είναι αυτοσωματική επικρατούσα, φυλοσύνδετη ή αυτοσωματική υπολειπόμενη. Η CMT σχετίζεται με περισσότερα από 80 αιτιολογικά γονίδια και χιλιάδες μεταλλάξεις.
Σήμερα, με την ραγδαία ανάπτυξη της τεχνολογίας και τη χρήση της τεχνολογίας αλληλούχισης επόμενης γενιάς (Next Generation Sequencing – NGS) ως διαγνωστικό εργαλείο, είναι δυνατή η ταχύτερη διάγνωση και η διάγνωση σπάνιων περιπτώσεων.
Το Τμήμα Νευρογενετικής του Ινστιτούτου Νευρολογίας και Γενετικής Κύπρου μελετά την πολυνευροπάθεια CMT για πάνω από 30 χρόνια συμβάλλοντας σημαντικά, τόσο στον τομέα των διαγνωστικών υπηρεσιών με μεγάλο αριθμό διαγνωστικών τεστ, όσο και στον τομέα της έρευνας με σημαντικά ευρήματα. Έχει διεξάγει πέραν των 800 διαγνωστικών εξετάσεων και έχει διαγνώσει πέραν των 200 ασθενών με πολυνευροπάθεια CMT. Μέσα από τα διαγνωστικά και ερευνητικά προγράμματα έχει ανιχνεύσει σημαντικό αριθμό καινούριων μεταλλάξεων σε γνωστά γονίδια συμπεριλαμβανομένων των γονιδίων PMP22, MPZ, GJB1 και MFN2 [1]. Χαρακτηριστική είναι η μετάλλαξη PMP22 S22F που έχει βρεθεί μόνο στον Κυπριακό πληθυσμό και σε πολύ ψηλό ποσοστό [2].
Τα τελευταία τέσσερα χρόνια το Τμήμα Νευρογενετικής ενέταξε το διαγνωστικό αυτό εργαλείο NGS για διερεύνηση των γονιδίων που σχετίζονται με τις πολυνευροπάθειες CMT. Με τη χρήση της τεχνολογίας NGS είναι δυνατή η αλληλούχιση μεγάλου μέρους του γονιδιώματος σε πολύ σύντομο χρόνο. Αυτό δίνει τη δυνατότητα διερεύνησης πολύ σπάνιων γονιδίων που προηγουμένως ήταν πολύ πιο δαπανηρή και χρονοβόρα. Με την εφαρμογή της τεχνολογίας NGS έχουν διαγνωστεί πέραν των 10 ασθενών με CMT (σε κάποιες περιπτώσεις ασθενείς αδιάγνωστοι για χρόνια), με ανεύρεση τόσο γνωστών όσο και νέων πιθανών υπεύθυνων μεταλλάξεων συμπεριλαμβανομένων των γονιδίων ATP1A1, GDAP1, MPZ, RAB7A, SPTLC1, JAG1, FBLN5, JAG1, ATP7A, MME και SCN10A.
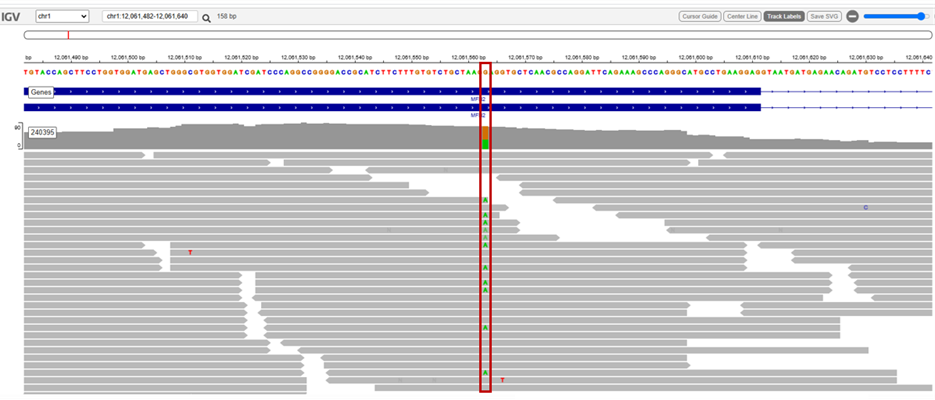
Εικόνα 1: Ενδεικτικό διάγραμμα αλληλούχισης (IGV) της μετάλλαξής MFN2:c.922G>A.
References:
- Nicolaou P, et.al. Charcot-Marie-Tooth disease in Cyprus: epidemiological, clinical and genetic characteristics. Neuroepidemiology. 2010;35(3):171-7.
- Kleopa KA, et.al. A novel PMP22 mutation Ser22Phe in a family with hereditary neuropathy with liability to pressure palsies and CMT1A phenotypes. Neurogenetics 2004; 5: 171–175.
- Cinarli F, et.al. The phenotypic spectrum of pathogenic ATP1A1 variants expands: the novel p.P600R substitution causes demyelinating Charcot-Marie-Tooth disease. J Neurol. 2023 May;270(5):2576-2590.
Δρ Πασχάλης Νικολάου
Scientist
Τμήμα Νευρογενετικής
Ινστιτούτο Νευρολογίας και Γενετικής Κύπρου






0002.jpg)