Cytogenetics and Genomics Department
ΥΠΗΡΕΣΙΕΣ, Γνωρίζοντας το Σύνδρομο Εύθραυστου Χ

Τo Σύνδρομο Fragile X περιεγράφηκε για πρώτη φορά το 1943 ως μία κατάσταση φυλοσύνδετης νοητικής αναπηρίας, συνήθως μέτριας μορφής στα αγόρια και ήπιας μορφής στα κορίτσια με συχνότητα εμφάνισης 1:4000 και 1:8000 αντίστοιχα. Θεωρείται η δεύτερη πιο συχνή αιτία νοητικής αναπηρίας σε αγόρια μετά το σύνδρομο Down και η πιο συχνή κληρονομική μορφή. Το σύνδρομο έγινε γνωστό ως το σύνδρομο εύθραυστου Χ επειδή ορισμένα άτομα με τη διαταραχή αυτή βρέθηκαν να έχουν ένα τμήμα του χρωμοσώματος Χ, το οποίο φαινόταν να είναι σπασμένο ή εύθραυστο, χωρίς να έχει αποχωριστεί όπως φαίνεται στην Εικόνα 1.

Μέσα από έρευνες βρέθηκε ότι το γονίδιο Fragile X Messenger Ribonucleoprotein 1 (FMR1) που παράγει την πρωτεΐνη FMRP, της οποίας η απουσία προκαλεί το σύνδρομο, βρίσκεται ακριβώς στο σημείο αυτό. Συγκεκριμένα το γονίδιο FMR1 βρίσκεται στην χρωμοσωμική περιοχή Χq27.3. Η κύρια παθογόνος μεταλλαγή του γονιδίου (99%) αφορά την εκτεταμένη επανάληψη τριπλέτας βάσεων CGG που προκαλεί την υπερμεθυλίωση του υποκινητή του γονιδίου FMR1 μειώνοντας ή αναστέλλοντας έτσι την παραγωγή της πρωτεΐνης FMRP.
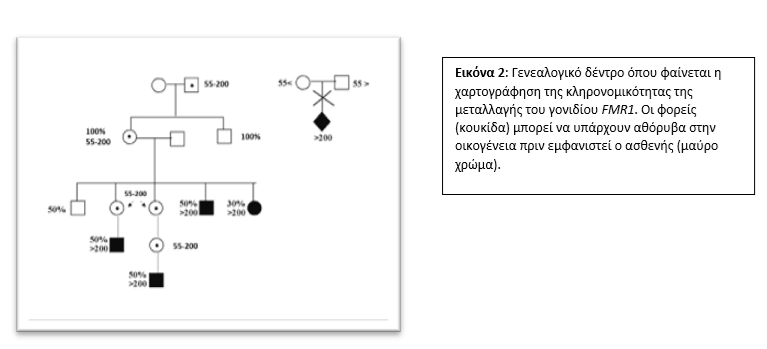
Ο φυσιολογικός μέγιστος αριθμός επαναλήψεων της CGG τριπλέτας έχει καθοριστεί ως ο 55 (<55). Εάν ένα άτομο φέρει από 55 μέχρι και 200 (55-200) επαναλήψεις τότε ονομάζεται φορέας του συνδρόμου και βρίσκεται σε μια κατάσταση προ-μεταλλαγής (premutation). Το άτομο που φέρει περισσότερες από 200 επαναλήψεις (>200) (full mutation) φέρει και το σύνδρομο του Εύθραυστου Χ. Εφόσον τα αγόρια φέρουν μόνο ένα Χ χρωμόσωμα (ΧΥ) και οι γυναίκες φέρουν δύο (ΧΧ), όταν ένας άντρας είναι φορέας της προ-μεταλλαγής ή είναι ασθενής με πλήρη μεταλλαγή θα κληρονομήσει υποχρεωτικά το παθολογικό Χ χρωμόσωμα στις κόρες του αλλά όχι στους γιους του [Εικ. 2]. Αντίστοιχα, εάν μία γυναίκα είναι φορέας προ-μεταλλαγής ή πλήρους μεταλλαγής, έχει πιθανότητα 50% να κληρονομήσει στα παιδιά της το παθολογικό Χ. Γι’ αυτό είναι σημαντικό μετά από κάθε διάγνωση να γίνεται περεταίρω διερεύνηση στην οικογένεια και ειδικά στις γυναίκες για σκοπούς πρόληψης.
Πολλές φορές η σοβαρότητα της κλινικής εικόνας ανάμεσα στις γενιές δεν είναι σταθερή. Τα κλινικά χαρακτηριστικά, του συνδρόμου συμπεριλαμβανομένου και της νοητικής αναπηρίας, εκτείνονται σε μεγάλο εύρος. Ο κύριος λόγος αφορά στον μηχανισμό επίσπευσης που μπορεί να μετατρέψει μια προ-μεταλλαγή σε πλήρη μεταλλαγή μέσα σε μια γενιά. Οι άντρες ασθενείς χαρακτηρίζονται από κάποια κύρια κλινικά χαρακτηριστικά όπως επίμηκες προσωπείο, ευμεγέθη και προεξέχοντα αυτιά, μακροορχιδισμό αλλά και νοητική αναπηρία. Οι γυναίκες φορείς, παρόλο που δεν ακολουθούν αυτά τα χαρακτηριστικά, μπορεί να παρουσιάζουν ψυχιατρικά και νευρολογικά προβλήματα και ελαφριά νοητική αναπηρία ή να έχουν φυσιολογικό κλινικό φαινότυπο. Η ένταση των συμπτωμάτων αυτών συνδέεται με το μοτίβο αδρανοποίησης του χρωμοσώματος Χ, φαινόμενο που αφορά την αδρανοποίηση ενός εκ των δύο χρωμοσωμάτων Χ σε κάθε κύτταρο της γυναίκας.
Άτομα με την προ-μεταλλαγή έχουν αυξημένο ρίσκο εμφάνισης μίας νευροεκφυλιστικής διαταραχής που ονομάζεται Fragile X tremor-ataxia syndrome (FXTAS) και παρουσιάζεται συνήθως στους άντρες και πιο σπάνια στις γυναίκες φορείς μετά την ηλικία των 60. Μια άλλη διαταραχή που εμφανίζεται σε γυναίκες με προ-μεταλλαγή είναι η Fragile X-associated primary ovarian insufficiency (FXPOI) και αφορά την πρόωρη εμμηνόπαυση σε ηλικία μικρότερη των 40.
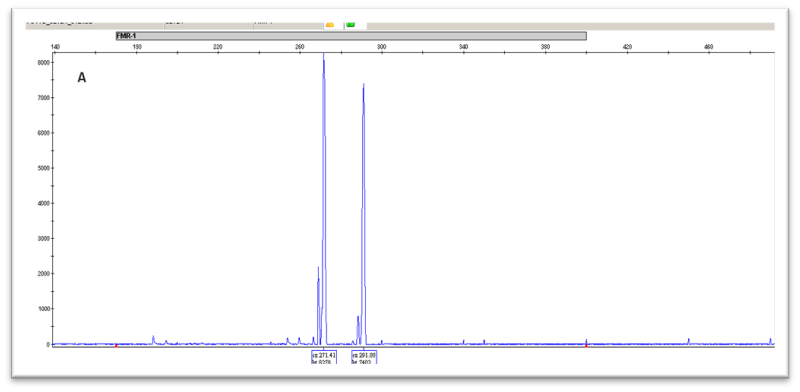
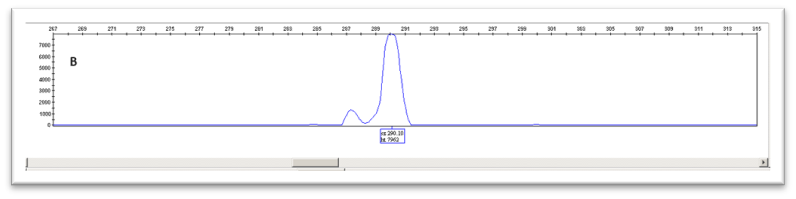
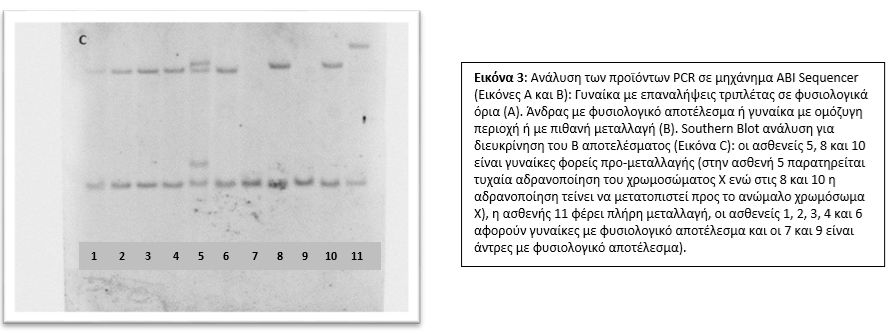
Στο Τμήμα Κυτταρογενετικής και Γονιδιωματικής προσφέρεται από το 1995 η γενετική διάγνωση περιστατικών Fragile-X για πλήρη μεταλλαγή (full mutation) ή προ-μεταλλαγή (premutation) μετά από παραπομπές από ειδικούς γιατρούς. Η διάγνωση γίνεται από εξειδικευμένο προσωπικό σε μεταγεννητικά δείγματα (δείγμα περιφερικού αίματος) και προγεννητικά δείγματα (δείγμα χορειακών λαχνών ή αμνιακού υγρού) (4-6). Η μεθοδολογία που ακολουθείται περιλαμβάνει μέθοδο PCR σε συνδυασμό με ανάλυση σε ABI Sequencer ή/και Southern Blot, υπολογίζοντας τον αριθμό των επαναλήψεων της τριπλέτας CGG καθορίζοντας έτσι το φάσμα του συνδρόμου στο οποίο βρίσκεται ο ασθενής [Εικ. 3]. Ακολουθεί συμβουλευτική από κλινικούς γενετιστές και η παρακολούθηση του ασθενούς από ομάδα ειδικών.
Βιβλιογραφία
1. V. Biancalana et al. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders, European Journal of Human Genetics (2015) 23, 417–425
2. W. Saldarriaga et al. Fragile X Syndrome, Saldarriaga W/et al/Colombia Médica (2014) 45, 4
3. C. Ciaccio et al, Fragile X syndrome: a review of clinical and molecular diagnoses, Italian Journal of Pediatrics (2017) 43:39
4. Patsalis PC et al. Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: Incidence, genetic variation and stability. American Journal of Medical Genetics (1999) 84:184-190
5. Patsalis PC et al. Cytogenetic and Fragile X Molecular Testing of Individuals with Mentally Retarded Population of Unknown Etiology. Genetic Counselling (1997) 8:1-6
6. Patsalis PC et al. Molecular Diagnosis and Frequency of the Fragile X Syndrome in the Cypriot Population. Genetic Counseling in the Down of the 21st Century, Editors Bartsokas SC and Beighton P, Zeta Medical Publications (1998), ISBN: 960-7144-45-7
Νικόλ Σαλάμε
Senior Laboratory Scientific Officer
Τμήμα Κυτταρογεντικής και Γονιδιωματικής






0002.jpg)