Blood Disorder Genetics and Thalassemia Department
ΕΡΕΥΝΑ - Νέα στρατηγική για τη β-θαλασσαιμία
 Μέλη της ομάδας της μελέτης και του Ινστιτούτου Νευρολογίας & Γενετικής. Από αριστερά προς δεξιά: Δρ. Carsten Werner Lederer, Δρ. Παναγιώτα Παπασάββα, Δρ. Νικολέττα Παπαϊωάννου, Δρ. Πέτρος Πατσαλή
Members of the study team and of Cyprus Institute of Neurology & Genetics. From left to right: Dr. Carsten Werner Lederer, Dr. Panayiota Papasavva, Dr. Nikoletta Papaioannou, Dr. Petros Patsali
Μέλη της ομάδας της μελέτης και του Ινστιτούτου Νευρολογίας & Γενετικής. Από αριστερά προς δεξιά: Δρ. Carsten Werner Lederer, Δρ. Παναγιώτα Παπασάββα, Δρ. Νικολέττα Παπαϊωάννου, Δρ. Πέτρος Πατσαλή
Members of the study team and of Cyprus Institute of Neurology & Genetics. From left to right: Dr. Carsten Werner Lederer, Dr. Panayiota Papasavva, Dr. Nikoletta Papaioannou, Dr. Petros Patsali
Μπορεί η διπλή γονιδιωματική επεξεργασία να επιτύχει θεραπευτική επαγωγή της εμβρυϊκής αιμοσφαιρίνης, διατηρώντας τη γονιδιωματική ακεραιότητα;
Η β-θαλασσαιμία, γνωστή και ως Μεσογειακή αναιμία, είναι ένα κληρονομικό νόσημα του αίματος και είναι αρκετά συχνό στην Κύπρο. Προκαλείται από μεταλλάξεις στο γονίδιο β (HBB), το οποίο είναι υπεύθυνο για την παραγωγή της β-σφαιρίνης, ενός βασικού συστατικού της ενήλικης αιμοσφαιρίνης (α2/β2). Η αιμοσφαιρίνη είναι μια πρωτεΐνη που βρίσκεται στα ερυθρά αιμοσφαίρια και μεταφέρει οξυγόνο σε όλο το σώμα. Όταν κληρονομηθούν δύο μεταλλαγμένα αντίγραφα του γονιδίου HBB, το γονίδιο δεν λειτουργεί σωστά, με αποτέλεσμα τη μειωμένη ή και πλήρη απουσία παραγωγής β-σφαιρίνης. Αυτό οδηγεί σε αναιμία και, συνεπώς, τα άτομα με β-θαλασσαιμία χρειάζονται τακτικές μεταγγίσεις αίματος σε όλη τη διάρκεια της ζωής τους.
Έχει αποδειχθεί ότι η αύξηση της γ-σφαιρίνης, η οποία αποτελεί συστατικό της εμβρυϊκής αιμοσφαιρίνης (α2/γ2, HbF), μπορεί να έχει θεραπευτική δράση, καθώς αντισταθμίζει την έλλειψη της β-σφαιρίνης. Η παραγωγή της εμβρυϊκής αιμοσφαιρίνης μειώνεται φυσιολογικά μετά τη γέννηση και αντικαθίσταται από την ενήλικη αιμοσφαιρίνη. Για να συμβεί αυτή η διαδικασία, γνωστή ως hemoglobin switching (μεταστροφή αιμοσφαιρίνης), εμπλέκονται διάφορα γονίδια και μοριακοί μηχανισμοί. Επομένως, αν καταφέρουμε να μειώσουμε τη δράση ορισμένων από αυτά τα γονίδια, μπορούμε να επανενεργοποιήσουμε την παραγωγή της HbF, οδηγώντας σε μια αποτελεσματική θεραπευτική προσέγγιση για τις β-αιμοσφαιρινοπάθειες. Τα τελευταία χρόνια, η γονιδιακή θεραπεία έχει σημειώσει ραγδαία πρόοδο και βρίσκεται όλο και πιο κοντά στην ανάπτυξη οριστικών θεραπειών. Σημαντικό ρόλο σε αυτή την εξέλιξη έχουν τα εργαλεία επεξεργασίας γονιδιώματος, όπως το CRISPR/Cas9 και το base editing (επεξεργασία βάσεων). Συγκεκριμένα, η τεχνολογία επεξεργασίας βάσεων επιτρέπει την ακριβή τροποποίηση συγκεκριμένων σημείων του DNA, αλλάζοντας μία βάση σε μία άλλη (C→T ή A→G). Με απλά λόγια, λειτουργεί σαν ένα «μολύβι με γόμα», που μπορεί να διορθώσει ένα «γράμμα» στον γενετικό κώδικα και να το αντικαταστήσει με ένα άλλο, οδηγώντας σε αλλαγές στη λειτουργία ή στην έκφραση ενός γονιδίου.
Στην πρόσφατη μελέτη μας, με τίτλο “Functional correction and genome integrity with duplex base editing of β-thalassemic hematopoietic stem cells”, που δημοσιεύθηκε στο διεθνές επιστημονικό περιοδικό Genome Biology, διερευνήσαμε πώς η τεχνολογία επεξεργασίας βάσεων μπορεί να αξιοποιηθεί για την αύξηση της εμβρυϊκής αιμοσφαιρίνης σε CD34+ προγονικά αιμοποιητικά βλαστοκύτταρα προερχόμενα από άτομα με β-θαλασσαιμία. Η ερευνητική μας προσπάθεια στο Τμήμα Γενετικής Αιματολογικών Νοσημάτων & Θαλασσαιμίας του Ινστιτούτου Νευρολογίας & Γενετικής Κύπρου (ΙΝΓΚ) πραγματοποιήθηκε σε συνεργασία με τις Κλινικές Θαλασσαιμίας Λευκωσίας και Λάρνακας του ΟΚΥπΥ και το Πανεπιστημιακό Ιατρικό Κέντρο του Freiburg στη Γερμανία.
Στη μελέτη μας αναπτύξαμε μια διπλή επεξεργασία βάσεων στρατηγική (2BE), στοχεύοντας ταυτόχρονα δύο γονίδια που εμπλέκονται στην επαγωγή της HbF, τον ερυθροειδή ενισχυτή του BCL11A και τον υποκινητή του γονιδίου γ (HBG). Προηγούμενες μελέτες έχουν δείξει ότι το γονίδιο BCL11A έχει σημαντική επίδραση στη φυσική μείωση της γ-σφαιρίνης, ενώ σημειακές μεταλλάξεις στον υποκινητή του HBG προκαλούν την κληρονομική επιμονή της εμβρυϊκής αιμοσφαιρίνης (HPFH), μια καλοήθη γενετική κατάσταση που χαρακτηρίζεται από συνεχιζόμενη παραγωγή υψηλών επιπέδων HbF.
Στόχος μας ήταν η ταυτόχρονη στόχευση των γονιδίων BCL11A και HBG ώστε να επιτευχθεί αύξηση της HbF σε θεραπευτικά επίπεδα. Για τον σκοπό αυτό, η διπλή επεξεργασία βάσεων στρατηγική συγκρίθηκε με προσεγγίσεις που στόχευαν μόνο το BCL11A (BE-BCL11A) ή μόνο το HBG (BE-HBG), καθώς και με την εγκεκριμένη από το FDA θεραπεία CASGEVY, η οποία βασίζεται στο CRISPR/Cas9 (DSB-Cas9) και στοχεύει μόνο το BCL11A. Το CRISPR/Cas9 προκαλεί διπλές θραύσεις στο DNA (DSBs), οι οποίες μπορεί να οδηγήσουν σε γονιδιωματική αστάθεια. Αντίθετα, η τεχνολογία επεξεργασίας βάσεων είναι ανεξάρτητη από DSBs και θεωρείται δυνητικά πιο ασφαλής προσέγγιση.
Μετά την επεξεργασία των CD34+ κυττάρων και την ερυθροειδική διαφοροποίησή τους, πραγματοποιήθηκαν αναλύσεις σε επίπεδο DNA, RNA και πρωτεΐνης. Σε επίπεδο DNA επιβεβαιώθηκε η επιτυχής επεξεργασία βάσεων στις στοχευμένες περιοχές των δύο γονιδίων. Η έκφραση της HbF αυξήθηκε σε όλα τα επεξεργασμένα κύτταρα σε σύγκριση με τα μη επεξεργασμένα δείγματα, ωστόσο μόνο η διπλή επεξεργασία βάσεων προσέγγιση οδήγησε σε ιδιαίτερα υψηλή έκφραση (56,68%) και σε στατιστικά σημαντική αύξηση κατά 6,4 φορές. Αντίστοιχη αύξηση παρατηρήθηκε και σε επίπεδο mRNA. Η πιθανή επίδραση των εργαλείων επεξεργασίας στο γονιδίωμα αξιολογήθηκε με τη μέθοδο CAST-seq. Τα αποτελέσματα έδειξαν ότι προσεγγίσεις που προκαλούν διπλές θραύσεις DNA, όπως το CRISPR/Cas9, μπορεί να συνοδεύονται από χρωμοσωμικές αναδιατάξεις. Αντίθετα, η τεχνολογία επεξεργασίας βάσεων, ακόμη και όταν εφαρμόζεται ταυτόχρονα σε δύο στόχους, μείωσε σημαντικά αυτούς τους κινδύνους. Ιδιαίτερα σημαντικό ήταν ότι δεν παρατηρήθηκε μετατόπιση μεταξύ των γονιδίων BCL11A και HBG μετά τη διπλή επεξεργασία.
Συνολικά, επιβεβαιώθηκε ότι η διπλή επεξεργασία βάσεων στρατηγική οδηγεί σε υψηλή επαγωγή της HbF, ξεπερνώντας το κλινικά σημαντικό όριο του 30%, ενώ παράλληλα διατηρεί υψηλό επίπεδο ασφάλειας. Τα ευρήματα αυτά αναδεικνύουν τη δυναμική της προσέγγισης ως υποσχόμενη θεραπευτική στρατηγική για τη β-θαλασσαιμία, αλλά και ως πιθανό μοντέλο εφαρμογής σε άλλες γενετικές ασθένειες.
Για περισσότερες πληροφορίες αναφορικά με τη μελέτη μπορείτε να επισκεφθείτε το δημοσιευμένο άρθρο ακολουθώντας τον παρακάτω σύνδεσμο: https://link.springer.com/article/10.1186/s13059-026-03974-7
Το έργο συγχρηματοδοτήθηκε από το έργο «Νέα Υποδομή για τη Διάγνωση και Θεραπεία Ασθενών» των Norway Grants 2014–2021, από το Ευρωπαϊκό Ταμείο Περιφερειακής Ανάπτυξης και την Κυπριακή Δημοκρατία, μέσω του Ιδρύματος Έρευνας και Καινοτομίας, στο πλαίσιο των προγραμμάτων EXCELLENCE/1216/0092 και EXCELLENCE/0421/0086, καθώς και από τη βασική χρηματοδότηση του Ινστιτούτου Νευρολογίας και Γενετικής Κύπρου. Η κινητικότητα και οι συνεργασίες για την παρούσα εργασία συγχρηματοδοτήθηκαν από τις Δράσεις COST CA21113 GenE-HumDi και CA22119 HELIOS, με την υποστήριξη του COST (European Cooperation in Science and Technology).
Θα θέλαμε να ευχαριστήσουμε τους συνεργάτες μας, τους φορείς χρηματοδότησης και το ΙΝΓΚ για τη σημαντική συμβολή τους στο έργο, καθώς και τα άτομα με β-θαλασσαιμία, χωρίς την προσφορά των οποίων η μελέτη αυτή δεν θα μπορούσε να πραγματοποιηθεί.
Δρ. Νικολέττα Παπαϊωάννου
RESEARCH
“Emerging gene editing strategy for β-Thalassemia”
Can duplex genome editing enable therapeutically relevant fetal hemoglobin induction while preserving genome integrity?
β-Thalassemia, also known as Mediterranean anemia, is an inherited blood disorder that is relatively common in Cyprus. It is caused by mutations in the β-globin gene (HBB), which is responsible for the production of β-globin, a key component of adult hemoglobin (α2/β2). Hemoglobin is a protein found in red blood cells that carries oxygen throughout the body. When two mutated copies of the HBB gene are inherited, the gene does not function properly, resulting in reduced or complete absence of β-globin production. This leads to anemia, and consequently individuals with β-thalassemia require regular blood transfusions throughout their lifetime.
It has been demonstrated that increasing the levels of γ-globin, a component of fetal hemoglobin (α2/γ2, HbF), can have therapeutic benefits, as it can compensate the defective β-globin. The production of fetal hemoglobin naturally declines after birth and is replaced by adult hemoglobin. This transition, known as hemoglobin switching, involves multiple genes and molecular regulatory mechanisms. Therefore, if the activity of specific genes involved in this process can be reduced, the production of HbF can be reactivated, potentially leading to an effective therapeutic strategy for β-hemoglobinopathies. In recent years, gene therapy has advanced rapidly and is moving closer to the development of routine treatments. Genome editing tools such as CRISPR/Cas9 and base editing have played a major role in this progress. In particular, base editing technology enables the precise modification of specific DNA sites by converting one nucleotide into another (C→T or A→G). In simple terms, it functions like a “pencil with an eraser,” capable of correcting a single “letter” in the genetic code, thereby altering gene function or expression.
In our recent study, entitled “Functional correction and genome integrity with duplex base editing of β-thalassemic hematopoietic stem cells,” published in the international scientific journal Genome Biology, we investigated how base editing technology can be used to increase fetal hemoglobin production in CD34+ hematopoietic stem and progenitor cells (HSPCs) derived from individuals with β-thalassemia. This research was conducted at the Department of Blood Disorder Genetics & Thalassemia (BDGT) of the Cyprus Institute of Neurology & Genetics (CING), in collaboration with the Thalassemia Clinics of Nicosia and Larnaca of the State Health Services Organisation, as well as the University Medical Center Freiburg in Germany.
In this study, we developed a duplex base editing strategy (2×BE) targeting simultaneously two genetic elements involved in HbF induction: the erythroid enhancer of BCL11A and the promoter region of the γ-globin gene (HBG). Previous studies have shown that BCL11A plays a major role in the physiological silencing of γ-globin expression, while point mutations in the HBG promoter are associated with hereditary persistence of fetal hemoglobin (HPFH), a benign genetic condition characterized by sustained high levels of HbF.
Our aim was therefore to simultaneously target BCL11A and HBG in order to achieve therapeutically relevant HbF induction. For this purpose, the duplex base editing approach was compared with single-target strategies focusing on either BCL11A (BE-BCL11A) or HBG (BE-HBG), as well as with the FDA-approved gene editing therapy CASGEVY, which is based on CRISPR/Cas9-mediated double-strand breaks (DSB-Cas9) and targets only BCL11A. CRISPR/Cas9 induces double-strand DNA breaks, which may lead to genomic instability. In contrast, base editing operates independently of DSB formation and is therefore considered a potentially safer genome editing approach.
Following genome editing of CD34+ cells and their subsequent differentiation towards the erythroid lineage, analyses were performed at the DNA, RNA, and protein levels. At the DNA level, efficient and precise base editing was confirmed at the targeted regions of both genes. HbF expression increased in all edited cells compared with unedited controls; however, the duplex base editing strategy resulted in the highest expression 56.86% and a statistically significant 6.4-fold increase. A corresponding increase was also observed at the mRNA level. The potential genomic impact of the editing tools was further assessed using CAST-Seq methodology. Results indicated that approaches involving DNA double-strand breaks, such as CRISPR/Cas9, may be associated with chromosomal rearrangements. In contrast, base editing, even when applied simultaneously at two genomic targets, significantly reduced these risks. Notably, no chromosomal translocation between BCL11A and HBG was detected following duplex base editing.
Overall, our findings demonstrate that the duplex base editing strategy can achieve robust induction of HbF above the clinically relevant therapeutic threshold of 30%, while maintaining a high safety profile. These results highlight the potential of this approach as a promising therapeutic strategy for β-thalassemia and suggest its broader applicability as a model for genome editing-based treatments of other genetic diseases.
For more information, the published article can be accessed at the following link:
https://link.springer.com/article/10.1186/s13059-026-03974-7
The project was co-funded by the project “New Infrastructure for the Diagnosis and Treatment of Patients” under the Norway Grants 2014–2021, by the European Regional Development Fund and the Republic of Cyprus, through the Research and Innovation Foundation, within the framework of the programmes EXCELLENCE/1216/0092 and EXCELLENCE/0421/0086, as well as by the core funding of the Cyprus Institute of Neurology and Genetics. The mobility and collaborations for this work were co-funded by COST Actions CA21113 GenE-HumDi and CA22119 HELIOS, with the support of COST (European Cooperation in Science and Technology).
Finally, we would like to sincerely thank our collaborators, funding bodies, and the CING for their valuable support, as well as the individuals with β-thalassemia whose contribution made this study possible.
Dr Nikoletta Papaioannou
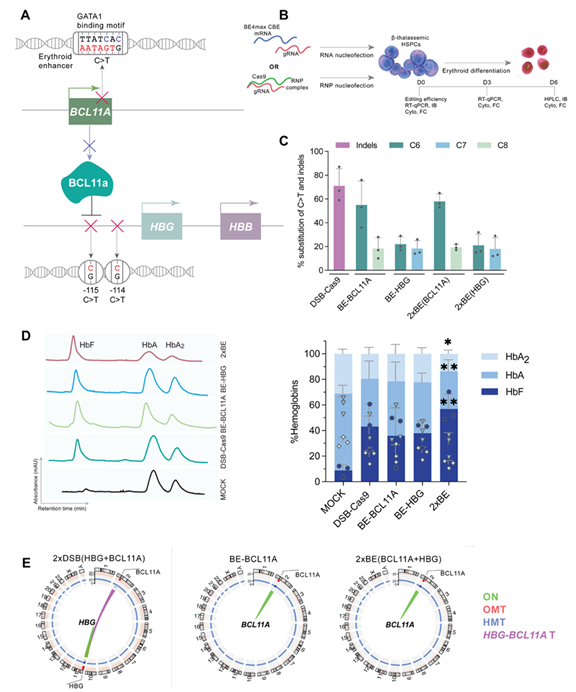
Σχεδιασμός της μελέτης και αποτελέσματα για τη διπλή επεξεργασία βάσεων (base editing) προσέγγιση. (A) Σχηματική απεικόνιση των στόχων για γονιδιακή επεξεργασία στα γονίδια HBG και BCL11A. (B) Σχηματική απεικόνιση της πειραματικής διαδικασίας. Τα εργαλεία επεξεργασίας γονιδιώματος (base editor & CRISPR/Cas9) εισήχθησαν με nucleofection σε β-θαλασσαιμικά προγονικά αιμοποιητικά βλαστοκύτταρα, ακολουθούμενη από ερυθροειδική διαφοροποίηση. Τα κύτταρα συλλέχθηκαν σε τρία διαφορετικά χρονικά σημεία που αντιστοιχούν σε πρώιμη (D0), ενδιάμεση (D3) και τελική (D6) φάση ερυθροποίησης, προκειμένου να πραγματοποιηθούν διάφορες αναλύσεις σε επίπεδο DNA, RNA και πρωτεΐνης. (C) Αποδοτικότητα επεξεργασίας DNA και αξιολόγηση indels. Τα αποτελέσματα της επεξεργασίας παρουσιάζονται ως ποσοστό (%) μετατροπής C>T για την επεξεργασία βάσεων προσέγγιση και ως ποσοστό (%) indels για το CRISPR/Cas9. (D) Λειτουργική αποκατάσταση μετά τη διπλή επεξεργασία βάσεων. Χρωματογραφήματα και ποσοτικοποίηση αποτελεσμάτων της HPLC analysis μετά από κάθε αντίστοιχη θεραπευτική προσέγγιση. Στα αριστερά παρουσιάζονται χρωματογραφήματα με κορυφές που αντιστοιχούν στις αιμοσφαιρίνες HbF, HbA και HbA2, ενώ στα δεξιά παρουσιάζεται η ποσοτικοποίηση των επιπέδων αιμοσφαιρίνης εκφρασμένων ως ποσοστό (%). (E) Δομικές γενετικές μεταβολές. Circos plots απεικονίζουν τις χρωμοσωμικές αναδιατάξεις που ανιχνεύθηκαν με την ανάλυση CAST-Seq. Η πράσινη γραμμή υποδηλώνει τον στόχο, ενώ η μωβ γραμμή υποδηλώνει τη μετατόπιση μεταξύ των δύο on-target θέσεων HBG και BCL11A (HBG-BCL11A T).
Study design and results for duplex base editing. (A) Schematic diagram illustrating the genome editing targets within the HBG and BCL11A genes. (B) A schematic diagram showing the experimental procedure. The base editor as mRNAs mixed with gRNA, or the DSB-based editor as a Cas9/gRNA RNP complex were nucleofected in β-thalassemic HSPCs followed by induction of erythroid differentiation. Cells were collected at three different time points representing early (D0), intermediate (D3) and late (D6) stages of erythropoiesis, to perform several analyses at DNA, RNA and protein level. (C) DNA editing efficiency and indel assessment. Editing efficiency results represented as % substitution of C>T for base editing and % indels for CRISPR/Cas9. (D) Functional correction after duplex base editing. Chromatograms and quantification of HPLC analysis results after each corresponding treatment. (left) Chromatograms with peaks presenting HbF, HbA and HbA2 hemoglobin. (right) Quantification of hemoglobin levels expressed as percentage (%). (E) Structural variations. Circos plots show the chromosomal rearrangements detected by CAST-Seq analysis. The green line shows the on-target (ON) and a purple line shows the translocation between the two on-target sites HBG and BCL11A (HBG-BCL11A T).





0002.jpg)