Molecular Genetics, Function & Therapy Department
ΜΕΛΟΣ ΣΤΟ Endo-ERN, Τμήμα Μοριακής Γενετικής Λειτουργίας & Θεραπείας

Το Τμήμα Μοριακής Γενετικής, Λειτουργίας και Θεραπείας (ΜΓΛΘ) του Ινστιτούτου Νευρολογίας και Γενετικής Κύπρου (ΙΝΓΚ), διαπιστευμένο με ISO 15189, λειτουργεί ως Κέντρο Αναφοράς του ευρωπαϊκού δικτύου Endo-ERN, προάγοντας τη φροντίδα υγείας και τη γρήγορη διάγνωση σπάνιων ενδοκρινικών παθήσεων. [1, 2].
Το ΜΓΛΘ διαθέτει μακρά εμπειρία στη διάγνωση και έρευνα κληρονομικών ενδοκρινικών παθήσεων από τη δεκαετία του 2000, χρησιμοποιώντας σύγχρονες τεχνολογίες για την ολοκληρωμένη διάγνωση ενδοκρινοπαθειών. Αυτές περιλαμβάνουν: τη Συγγενή Υπερπλασία Επινεφριδίων (Congenital Adrenal Hyperplasia [CAH]) [3, 4], τις διαταραχές της διαφοροποίησης του φύλου (Disorders of Sexual Differentiation) [5], τον τύπο 2A και 2B του συνδρόμου πολλαπλών ενδοκρινών νεοπλασιών (Multiple Endocrine Neoplasia) [6], γενετικές καταστάσεις που προκαλούν υπογοναδοτροπικό υπογοναδισμό [7], πρώιμη και καθυστερημένη εφηβεία [8], νεογνικό διαβήτη τύπου MODY [9], περιπτώσεις παχυσαρκίας και αρκετές άλλες σπανιότερες διαταραχές [10].
1. Διάγνωση Συγγενούς Υπερπλασίας Επινεφριδίων (Congenital Adrenal Hyperplasia - CAH) λόγω παθογόνων μεταλλάξεων στο γονίδιο CYP21A2
Η κλινική εικόνα της CAH παρουσιάζει μεγάλη ποικιλομορφία και οφείλεται σε γενετικές μεταλλάξεις στο γονίδιο CYP21A2. Η νόσος κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο. Οι πιο σοβαρές μορφές της, όπως η κλασική με απώλεια άλατος (Salt Wasting-SW) και η απλή αρρενοποιητική (Simple Virilizing-SV), επηρεάζουν περίπου 1 στα 10.000-20.000 άτομα [3, 11]. Αντίθετα, η μη κλασική CAH είναι μια πιο ήπια φαινοτυπική έκφραση της CAH, και οφείλεται σε σύνθετη ετεροζυγωτία ήπιων παθογόνων παραλλαγών στο γονίδιο CYP21A2 [12]. Η συχνότητα της νόσου εκτιμάται σε 1:100-1:500 γεννήσεων, με τα πιο συχνά συμπτώματα να είναι η υπερανδρογοναιμία και οι διαταραχές της γονιμότητας [12, 13]. Θηλυκά κυρίως άτομα, αλλά και άρρενες με ήπια μη κλασική μορφή εμφανίζουν σύνθετη ετεροζυγωτία δύο ήπιων παθογόνων παραλλαγών στο CYP21A2, με συμπτώματα υπερανδρογοναιμίας, υπερτρίχωσης, πρώιμης ήβης και υπογονιμότητας. Μελέτες, περιλαμβανομένης της έρευνας του ΜΓΛΘ, εκτιμούν τη συχνότητα φορέων των μεταλλάξεων CYP21A2 σε 1:10-1:25 στον γενικό πληθυσμό [13]. Η αναγνώριση ετερόζυγων φορέων στο CYP21A2 μπορεί να γίνει μέσω της μέτρησης της ACTH-διεγερμένης 17-ΟΗ προγεστερόνης (17-OHP). Ωστόσο, αυτή η συγκεκριμένη δοκιμή παρουσιάζει μεγαλύτερη επικάλυψη με τον υγιή πληθυσμό. Πρόσφατες μελέτες από το τμήμα ΜΓΛΘ έχουν δείξει ότι ένα σημαντικό ποσοστό γυναικών ετεροζυγωτών στο CYP21A2 ενδέχεται να διατρέχουν αυξημένο κίνδυνο εμφάνισης υπερανδρογοναιμίας [14-16]. Από το 2006, έχουν πραγματοποιηθεί πάνω από 1200 διαγνωστικές γονοτυπικές αναλύσεις για το γονίδιο CYP21A2 (Εικ. 1). Οι γενετικές αναλύσεις για τη CAH προέρχονταν κυρίως από κρατικά νοσοκομεία της Κύπρου, με λιγότερες παραπομπές από τον ιδιωτικό τομέα. Οι περισσότερες αφορούσαν γυναίκες με υπερανδρογονισμό στην προεφηβική ή εφηβική ηλικία, με συμπτώματα όπως πρώιμη ηβική τριχοφυΐα, εξέλιξη οστικής ηλικίας, σοβαρή ακμή, υπερτρίχωση, διαταραχές εμμήνου ρύσεως και αυξημένα επίπεδα 17-OHP. Η συνεργασία οδήγησε σε σημαντικές δημοσιεύσεις, που ανέδειξαν τη συχνότητα του γονιδίου CYP21A2 στον κυπριακό πληθυσμό και παρείχαν πληροφορίες για την πολυαλληλία και το σύμπλεγμα RCCX στην περιοχή MHC III του χρωμοσώματος 6p21.3. (Εικ. 2) [17, 18].
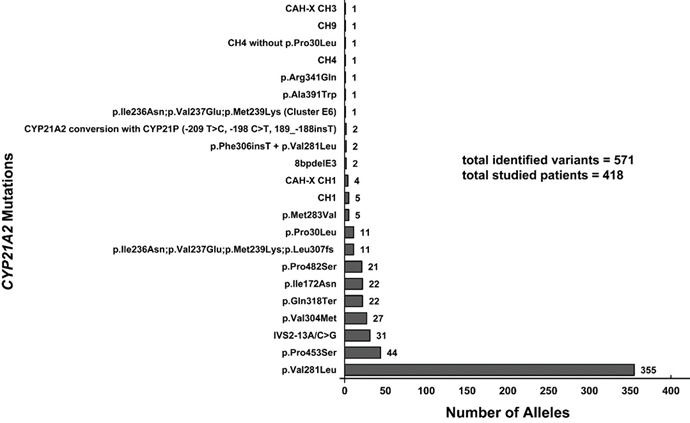
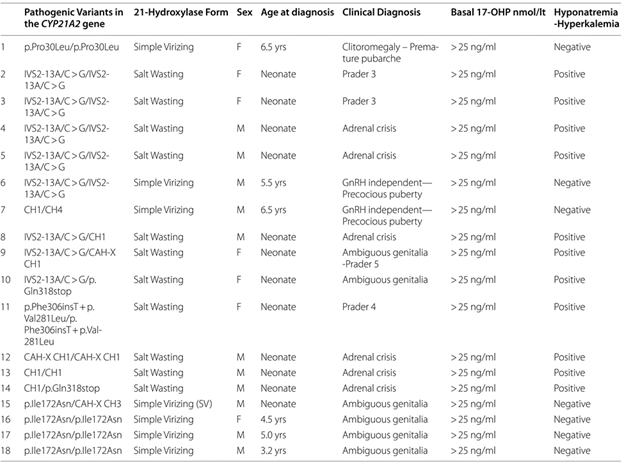
 Σύμφωνα με τις πρόσφατες κατευθυντήριες οδηγίες βέλτιστης πρακτικής για τη CAH [19], το ΜΓΛΘ χρησιμοποιεί κατά κόρον για τη γενετική διερεύνηση του γονιδίου CYP21A2 το συνδυασμό των μεθόδων Sanger sequencing και MLPA. Η MPLA είναι η πλέον κατάλληλη μέθοδος για την ανίχνευση σπάνιων διπλασιασμών/απαλείψεων στο γονίδιο CYP21A2. Το ΜΓΛΘ εμπλούτισε τη διερεύνηση του CYP21A2 με τη χρήση του ενζύμου TaqI, το οποίο ανιχνεύει χιμαιρικούς συνδυασμούς μεταξύ CYP21A1P και CYP21A2. Μέχρι σήμερα, έχουν εντοπιστεί 571 παθογόνες μεταλλάξεις στο CYP21A2, με την ήπια p.Val281Leu να είναι η πιο συχνή (62.17%) (Εικ. 3) [1]. Η πρόσφατη ανάλυση του επιπολασμού των αλληλόμορφων CYP21A2 σε Κύπριους ασθενείς κατέγραψε 265 ετεροζυγώτες και 153 σύνθετους ετεροζυγώτες/ομοζυγώτες. Συνολικά, 18 Κύπριοι ασθενείς έχουν γεννηθεί με την κλασική μορφή της CAH, με λεπτομερή συσχέτιση γονότυπου/φαινοτύπου να παρουσιάζεται στον Πίνακα 1. Παθογόνες παραλλαγές του CYP21A2 έχουν μελετηθεί εκτενώς, επιβεβαιώνοντας τους κλινικούς φαινότυπους στους Κύπριους ασθενείς. Το ΜΓΛΘ συνεργάζεται στενά με το δίκτυο ENDO-ERN, κοινοποιώντας τακτικά ευρήματα μέσω σεμιναρίων, συναντήσεων και της πλατφόρμας EuRRECa (https://endo-ern.eu/registries/eurreca/).
Σύμφωνα με τις πρόσφατες κατευθυντήριες οδηγίες βέλτιστης πρακτικής για τη CAH [19], το ΜΓΛΘ χρησιμοποιεί κατά κόρον για τη γενετική διερεύνηση του γονιδίου CYP21A2 το συνδυασμό των μεθόδων Sanger sequencing και MLPA. Η MPLA είναι η πλέον κατάλληλη μέθοδος για την ανίχνευση σπάνιων διπλασιασμών/απαλείψεων στο γονίδιο CYP21A2. Το ΜΓΛΘ εμπλούτισε τη διερεύνηση του CYP21A2 με τη χρήση του ενζύμου TaqI, το οποίο ανιχνεύει χιμαιρικούς συνδυασμούς μεταξύ CYP21A1P και CYP21A2. Μέχρι σήμερα, έχουν εντοπιστεί 571 παθογόνες μεταλλάξεις στο CYP21A2, με την ήπια p.Val281Leu να είναι η πιο συχνή (62.17%) (Εικ. 3) [1]. Η πρόσφατη ανάλυση του επιπολασμού των αλληλόμορφων CYP21A2 σε Κύπριους ασθενείς κατέγραψε 265 ετεροζυγώτες και 153 σύνθετους ετεροζυγώτες/ομοζυγώτες. Συνολικά, 18 Κύπριοι ασθενείς έχουν γεννηθεί με την κλασική μορφή της CAH, με λεπτομερή συσχέτιση γονότυπου/φαινοτύπου να παρουσιάζεται στον Πίνακα 1. Παθογόνες παραλλαγές του CYP21A2 έχουν μελετηθεί εκτενώς, επιβεβαιώνοντας τους κλινικούς φαινότυπους στους Κύπριους ασθενείς. Το ΜΓΛΘ συνεργάζεται στενά με το δίκτυο ENDO-ERN, κοινοποιώντας τακτικά ευρήματα μέσω σεμιναρίων, συναντήσεων και της πλατφόρμας EuRRECa (https://endo-ern.eu/registries/eurreca/).
2. Διάγνωση Πολλαπλής Ενδοκρινικής Νεοπλασίας τύπου 2 στην Κύπρο: Ο ρόλος των παθογόνων
ελαττωμάτων στο πρωτο-ογκογονίδιο RET
Το σύνδρομο Πολλαπλής Ενδοκρινικής Νεοπλασίας τύπου 2 (MEN2) είναι μια σπάνια, κληρονομική διαταραχή που προκαλεί ανάπτυξη όγκων σε ορμονοπαραγωγούς αδένες (θυρεοειδής, παραθυρεοειδής, επινεφρίδια) [20]. Κληρονομείται αυτοσωμικά επικρατητικά και συνδέεται με μυελικό καρκίνωμα του θυρεοειδούς (MTC), φαιοχρωμοκύτωμα και υπερπαραθυρεοειδισμό. Οι τρεις μορφές του (MEN2A, MEN2B, FMTC) εξαρτώνται από μεταλλάξεις στο γονίδιο RET, που ρυθμίζει την κυτταρική ανάπτυξη [21]. Οι μεταλλάξεις ταξινομούνται από την American Thyroid Association (ATA) και τη European Thyroid Association (ETA) σε τρεις κατηγορίες κινδύνου: υψηλότερο (ATA-HST), υψηλό (ATA-H) και μέτριο (ΑΤΑ-ΜΟD), με τις περισσότερες να εντοπίζονται στα εξόνια 10, 11, 13, 14, 15 και 16 [21, 22].
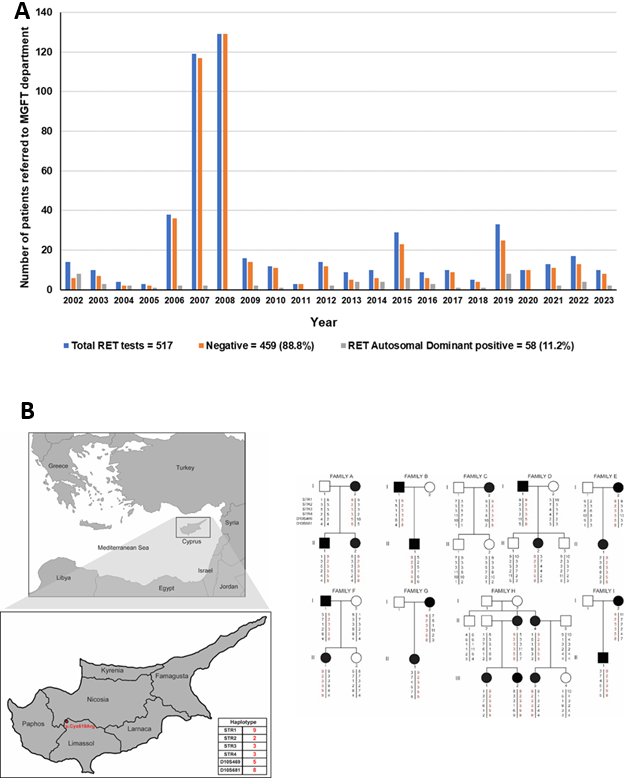
 Από το 2002, το τμήμα ΜΓΛΘ ανέλυσε το γονίδιο RET σε πάνω απο 500 ασθενείς με υποψία MEN2, ανιχνεύοντας παθογόνες παραλλαγές στο 11.2% (58 άτομα) [6, 23]. Από 22 μη συγγενείς οικογένειες, οι 12 (54.5%) έφεραν την παραλλαγή p.Cys618Arg, πιθανώς λόγω του φαινομένου του ιδρυτή (Founder effect) στην Κύπρο. Άλλες παραλλαγές περιλαμβάνουν τις p.Cys634Tyr, p.Val804Met, και p.Met918Thr [12] (Εικ. 4). Η έγκαιρη διάγνωση και η προληπτική θυρεοειδεκτομή, σύμφωνα με τις οδηγίες της ATA, είναι κρίσιμες για την πρόληψη του MTC σε ασθενείς και συγγενείς, παρέχοντας διαστρωμάτωση κινδύνου και στρατηγικές θεραπείας βασισμένες στις πιο πρόσφατες έρευνες.
Από το 2002, το τμήμα ΜΓΛΘ ανέλυσε το γονίδιο RET σε πάνω απο 500 ασθενείς με υποψία MEN2, ανιχνεύοντας παθογόνες παραλλαγές στο 11.2% (58 άτομα) [6, 23]. Από 22 μη συγγενείς οικογένειες, οι 12 (54.5%) έφεραν την παραλλαγή p.Cys618Arg, πιθανώς λόγω του φαινομένου του ιδρυτή (Founder effect) στην Κύπρο. Άλλες παραλλαγές περιλαμβάνουν τις p.Cys634Tyr, p.Val804Met, και p.Met918Thr [12] (Εικ. 4). Η έγκαιρη διάγνωση και η προληπτική θυρεοειδεκτομή, σύμφωνα με τις οδηγίες της ATA, είναι κρίσιμες για την πρόληψη του MTC σε ασθενείς και συγγενείς, παρέχοντας διαστρωμάτωση κινδύνου και στρατηγικές θεραπείας βασισμένες στις πιο πρόσφατες έρευνες.
3. Πρώιμη Εφηβεία: Γενετική Διερεύνηση σε μέλη οικογενειών με ιστορικό Κεντρικής Πρώϊμης Εφηβείας
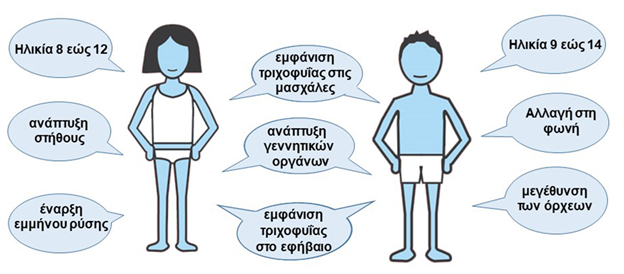
Η φυσιολογική εφηβεία ξεκινά στις ηλικίες 8-13 για τα κορίτσια και 9-14 για τα αγόρια, συνοδευόμενη από συναισθηματικές, ορμονικές και σωματικές αλλαγές, όπως θηλαρχή, εμμηναρχή, αλλαγές φωνής, μεγέθυνση όρχεων και ανάπτυξη τριχοφυΐας (Εικ. 5). Πρώιμη ήβη ορίζεται η έναρξη πριν τα 8 έτη στα κορίτσια και πριν τα 9 στα αγόρια. Η Κεντρική Πρώιμη Εφηβεία (ΚΠΕ) προκαλείται από πρόωρη ενεργοποίηση του άξονα υποθαλάμου-υπόφυσης-γονάδων (H-P-G), με έκκριση της εκλυτικής ορμόνης των γοναδοτροπινών (GnRH) από τον υποθάλαμο, που διεγείρει την παραγωγή ωχρινοποιητικής ορμόνης (LH) και ωοθυλακιοτρόπου ορμόνης (FSH) από την υπόφυση, ενεργοποιώντας τις γονάδες για παραγωγή οιστρογόνων και τεστοστερόνης.
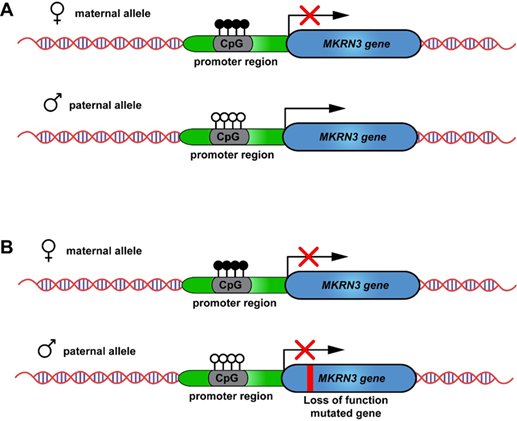
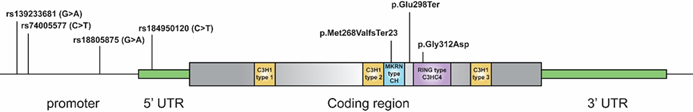
Ερευνητές του ΜΓΛΘ, σε συνεργασία με εξωτερικούς κλινικούς συνεργάτες, εντόπισαν το γονίδιο MKRN3 ως κρίσιμο παράγοντα για την ΚΠΕ, ιδιαίτερα σε γυναίκες Kυπριακής καταγωγής [8, 24-27]. Το MKRN3 ρυθμίζει τον χρόνο έναρξης της εφηβείας, και οι παθογόνες παραλλαγές του, που κληρονομούνται πατρικά (paternally inherited), αποτελούν κύρια αιτία ΚΠΕ, κυρίως στα κορίτσια. [28] (Εικ. 6). Μέχρι σήμερα, έχουν μελετηθεί περισσότερες από 100 Κύπριες με κλινική διάγνωση ΚΠΕ εντοπίζοντας παθογόνες παραλλαγές όχι μόνο στην περιοχή κωδικοποίησης πρωτεΐνης του MKRN3, αλλά και στον υποκινητή (promoter) και την περιοχή 5'-UTR.[8, 26] (Εικ. 7). Δώδεκα κορίτσια από 8 μη συγγενείς κυπριακές οικογένειες με ΚΠΕ παρουσίαζαν την ίδια παθογόνο παραλλαγή p.Gly312Asp, που προκαλεί απώλεια λειτουργίας, υποδηλώνοντας φαινόμενο ιδρυτή (founder effect). Αυτό συμβαίνει όταν νέος πληθυσμός προκύπτει από περιορισμένο αριθμό ατόμων, τροποποιώντας τη γενετική δομή και αυξάνοντας τη συχνότητα σπάνιων μεταλλάξεων, με αποτέλεσμα μεγαλύτερη πιθανότητα κληρονομικών ασθενειών [8, 24].
4. Καθυστερημένη Εφηβεία: Γενετική Διερεύνηση σε ασθενείς με οικογενειακό ιστορικό
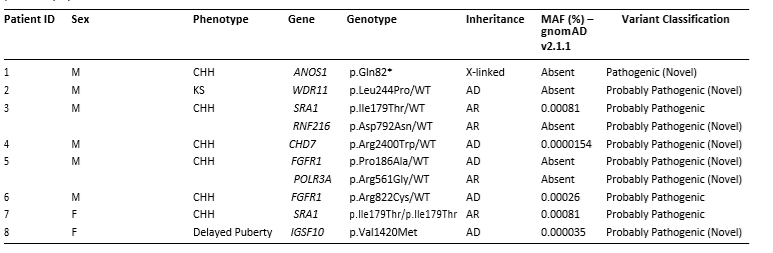
Ο Συγγενής Υπογοναδοτροπικός Υπογοναδισμός (ΣΥΥ) είναι σπάνια γενετική διαταραχή που επηρεάζει την ανάπτυξη της εφηβείας και τη γονιμότητα, προκαλώντας ανεπάρκεια των γονάδων, καθυστέρηση της εφηβείας και προβλήματα γονιμότητας. [29, 30]. Μέχρι τώρα, 8 άτομα (6 άνδρες και 2 γυναίκες) με ΣΥΥ διαγνώστηκαν μέσω Whole Exome Sequencing στο τμήμα ΜΓΛΘ του ΙΝΓΚ. Η ανάλυση αποκάλυψε μεταλλάξεις σε γονίδια σχετιζόμενα με ΣΥΥ και το σύνδρομο Kallmann, μια μορφή ΣΥΥ που περιλαμβάνει ανοσμία και καθυστερημένη εφηβεία (Πίνακας 2).[7]. Η μελέτη εντόπισε εννέα παθογόνες παραλλαγές που σχετίζονται με ΣΥΥ, επτά εκ των οποίων ήταν νέες. Υπολογιστική και δομική ανάλυση έδειξαν ότι αυτές οι μεταλλάξεις μπορεί να διαταράσσουν τη λειτουργία των πρωτεϊνών, συμβάλλοντας στην πρόκληση ΣΥΥ [7]. Οι έρευνες σε Κύπριους ασθενείς με ΣΥΥ ενισχύουν τη διαγνωστική ακρίβεια και μπορεί να οδηγήσουν σε νέες θεραπευτικές επιλογές.
5. Διαβήτης Ωριµότητας σε Νεαρά Άτοµα (Maturity Onset Diabetes of the Young): Γενετική Διερεύνηση
Το Τμήμα ΜΓΛΘ προσφέρει γενετικές εξετάσεις για τη διάγνωση και τον τύπο του μονογονιδιακού διαβήτη MODY, επιτρέποντας την ανάπτυξη εξατομικευμένων θεραπευτικών σχεδίων. Οι ασθενείς με MODY παρουσιάζουν προβλήματα γλυκόζης και ινσουλίνης σε νεαρή ηλικία και συχνά διαγιγνώσκονται λανθασμένα με διαβήτη τύπου 1 ή 2. Η μοριακή διερεύνηση γίνεται μέσω Whole Exome Sequencing (NGS), με ανίχνευση παθογόνων μεταλλάξεων σε 6 από 50 ασθενείς. Βρέθηκαν 4 μεταλλάξεις στο γονίδιο GCK (MODY2) και 2 στο γονίδιο HNF1α (MODY3). [9, 31]. Επιπλέον, έχουν εντοπιστεί άλλες 2 μεταλλάξεις στο σπάνιο γονιδίο ADCY3 σε πέντε διαφορετικούς ασθενείς με επιβεβαιωμένη διάγνωση παχυσαρκίας πρώιμης έναρξης [10].
Kαταληκτικές Παρατηρήσεις
Με 15 χρόνια έρευνας και διαγνωστικών υπηρεσιών για κληρονομικά ενδοκρινικά νοσήματα, το τμήμα ΜΓΛΘ του ΙΝΓΚ, ως κέντρο αναφοράς ENDO-ERN, έχει συγκεντρώσει πολύτιμα δεδομένα. Αυτά ενισχύουν τους στόχους των Ευρωπαϊκών Δικτύων Αναφοράς (ERNs) για τη βελτίωση της διάγνωσης και θεραπείας, συμβάλλοντας στην ποιοτική φροντίδα των ασθενών.
Δρ Βάσος Νεοκλέους
Scientist
Τμήμα Μοριακής Γενετικής, Λειτουργίας και Θεραπείας
Ινστιτούτο Νευρολογίας και Γενετικής Κύπρου
Email: [email protected]
Δρ Παύλος Φάνης
Associate Scientist
Τμήμα Μοριακής Γενετικής, Λειτουργίας και Θεραπείας
Ινστιτούτο Νευρολογίας και Γενετικής Κύπρου
Email: [email protected]
References
- Neocleous V, Fanis P, Toumba M, Skordis N, Phylactou LA: Genetic diagnosis of endocrine disorders in Cyprus through the Cyprus Institute of Neurology and Genetics: an ENDO-ERN Reference Center. Orphanet J Rare Dis 2024, 19(1):167.
- Eggermann T, Elbracht M, Kurth I, Juul A, Johannsen TH, Netchine I, Mastorakos G, Johannsson G, Musholt TJ, Zenker M et al: Genetic testing in inherited endocrine disorders: joint position paper of the European reference network on rare endocrine conditions (Endo-ERN). Orphanet J Rare Dis 2020, 15(1):144.
- Neocleous V, Fanis P, Toumba M, Stylianou C, Picolos M, Andreou E, Kyriakou A, Iasonides M, Nicolaou S, Kyriakides TC et al: The Spectrum of Genetic Defects in Congenital Adrenal Hyperplasia in the Population of Cyprus: A Retrospective Analysis. Horm Metab Res 2019, 51(9):586-594.
- Fanis P, Neocleous V, Kosta K, Karipiadou A, Hartmann MF, Wudy SA, Karantaglis N, Papadimitriou DT, Skordis N, Tsikopoulos G et al: Late diagnosis of 3beta-Hydroxysteroid dehydrogenase deficiency: the pivotal role of gas chromatography-mass spectrometry urinary steroid metabolome analysis and a novel homozygous nonsense mutation in the HSD3B2 gene. J Pediatr Endocrinol Metab 2021, 34(1):131-136.
- Neocleous V, Sismani C, Shammas C, Efstathiou E, Alexandrou A, Ioannides M, Argyrou M, Patsalis PC, Phylactou LA, Skordis N: Duplication of exons 3-10 of the HSD17B3 gene: a novel type of genetic defect underlying 17beta-HSD-3 deficiency. Gene 2012, 499(2):250-255.
- Fanis P, Skordis N, Frangos S, Christopoulos G, Spanou-Aristidou E, Andreou E, Manoli P, Mavrommatis M, Nicolaou S, Kleanthous M et al: Multiple endocrine neoplasia 2 in Cyprus: evidence for a founder effect. J Endocrinol Invest 2018, 41(10):1149-1157.
- Neocleous V, Fanis P, Toumba M, Tanteles GA, Schiza M, Cinarli F, Nicolaides NC, Oulas A, Spyrou GM, Mantzoros CS et al: GnRH Deficient Patients With Congenital Hypogonadotropic Hypogonadism: Novel Genetic Findings in ANOS1, RNF216, WDR11, FGFR1, CHD7, and POLR3A Genes in a Case Series and Review of the Literature. Frontiers in endocrinology 2020, 11:626.
- Neocleous V, Fanis P, Toumba M, Gorka B, Kousiappa I, Tanteles GA, Iasonides M, Nicolaides NC, Christou YP, Michailidou K et al: Pathogenic and Low-Frequency Variants in Children With Central Precocious Puberty. Frontiers in endocrinology 2021, 12:745048.
- Shammas C, Neocleous V, Phelan MM, Lian LY, Skordis N, Phylactou LA: A report of 2 new cases of MODY2 and review of the literature: implications in the search for type 2 diabetes drugs. Metabolism 2013, 62(11):1535-1542.
- Toumba M, Fanis P, Vlachakis D, Neocleous V, Phylactou LA, Skordis N, Mantzoros CS, Pantelidou M: Molecular modelling of novel ADCY3 variant predicts a molecular target for tackling obesity. International journal of molecular medicine 2022, 49(1).
- Auer MK, Nordenstrom A, Lajic S, Reisch N: Congenital adrenal hyperplasia. Lancet 2023, 401(10372):227-244.
- Neocleous V, Fanis P, Phylactou LA, Skordis N: Genotype Is Associated to the Degree of Virilization in Patients With Classic Congenital Adrenal Hyperplasia. Frontiers in endocrinology 2018, 9:733.
- Phedonos AA, Shammas C, Skordis N, Kyriakides TC, Neocleous V, Phylactou LA: High carrier frequency of 21-hydroxylase deficiency in Cyprus. Clin Genet 2013, 84(6):585-588.
- Neocleous V, Shammas C, Phedonos AA, Phylactou LA, Skordis N: Phenotypic variability of hyperandrogenemia in females heterozygous for CYP21A2 mutations. Indian J Endocrinol Metab 2014, 18(Suppl 1):S72-79.
- Skordis N, Shammas C, Phedonos AA, Kyriakou A, Toumba M, Neocleous V, Phylactou LA: Genetic defects of the CYP21A2 gene in girls with premature adrenarche. J Endocrinol Invest 2015, 38(5):535-539.
- Neocleous V, Fanis P, Toumba M, Phedonos AAP, Picolos M, Andreou E, Kyriakides TC, Tanteles GA, Shammas C, Phylactou LA et al: Variations in the 3'UTR of the CYP21A2 Gene in Heterozygous Females with Hyperandrogenaemia. International journal of endocrinology 2017, 2017:8984365.
- Fanis P, Skordis N, Toumba M, Picolos M, Tanteles GA, Neocleous V, Phylactou LA: The pathogenic p.Gln319Ter variant is not causing congenital adrenal hyperplasia when inherited in one of the duplicated CYP21A2 genes. Frontiers in endocrinology 2023, 14:1156616.
- Fanis P, Skordis N, Phylactou LA, Neocleous V: Salt-wasting congenital adrenal hyperplasia phenotype as a result of the TNXA/TNXB chimera 1 (CAH-X CH-1) and the pathogenic IVS2-13A/C > G in CYP21A2 gene. Hormones (Athens) 2023, 22(1):71-77.
- Baumgartner-Parzer S, Witsch-Baumgartner M, Hoeppner W: EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur J Hum Genet 2020, 28(10):1341-1367.
- Sahakian N, Castinetti F, Romanet P, Reznik Y, Brue T: Updates on the genetics of multiple endocrine neoplasia. Ann Endocrinol (Paris) 2024, 85(2):127-135.
- Wells SA, Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F et al: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25(6):567-610.
- Elisei R, Alevizaki M, Conte-Devolx B, Frank-Raue K, Leite V, Williams GR: 2012 European thyroid association guidelines for genetic testing and its clinical consequences in medullary thyroid cancer. European thyroid journal 2013, 1(4):216-231.
- Neocleous V, Fanis P, Frangos S, Skordis N, Phylactou LA: RET Proto-Oncogene Variants in Patients with Medullary Thyroid Carcinoma from the Mediterranean Basin: A Brief Report. Life (Basel) 2023, 13(6).
- Neocleous V, Shammas C, Phelan MM, Nicolaou S, Phylactou LA, Skordis N: In silico analysis of a novel MKRN3 missense mutation in familial central precocious puberty. Clin Endocrinol (Oxf) 2016, 84(1):80-84.
- Skordis N, Ferrari E, Antoniadou A, Phylactou LA, Fanis P, Neocleous V: GnRH-dependent precocious puberty manifested at the age of 14 months in a girl with 47,XXX karyotype. Hormones (Athens) 2017, 16(3):318-321.
- Fanis P, Skordis N, Toumba M, Papaioannou N, Makris A, Kyriakou A, Neocleous V, Phylactou LA: Central Precocious Puberty Caused by Novel Mutations in the Promoter and 5'-UTR Region of the Imprinted MKRN3 Gene. Frontiers in endocrinology 2019, 10:677.
- Fanis P, Morrou M, Tomazou M, Michailidou K, Spyrou GM, Toumba M, Skordis N, Neocleous V, Phylactou LA: Methylation status of hypothalamic Mkrn3 promoter across puberty. Frontiers in endocrinology 2022, 13:1075341.
- Tinano FR, Canton APM, Montenegro LR, de Castro Leal A, Faria AG, Seraphim CE, Brauner R, Jorge AA, Mendonca BB, Argente J et al: Clinical and Genetic Characterization of Familial Central Precocious Puberty. J Clin Endocrinol Metab 2023, 108(7):1758-1767.
- Vezzoli V, Hrvat F, Goggi G, Federici S, Cangiano B, Quinton R, Persani L, Bonomi M: Genetic architecture of self-limited delayed puberty and congenital hypogonadotropic hypogonadism. Frontiers in endocrinology 2022, 13:1069741.
- Al Sayed Y, Howard SR: Panel testing for the molecular genetic diagnosis of congenital hypogonadotropic hypogonadism - a clinical perspective. Eur J Hum Genet 2023, 31(4):387-394.
- Neocleous V, Shammas C, Phelan MM, Fanis P, Pantelidou M, Skordis N, Mantzoros C, Phylactou LA, Toumba M: A novel MC4R deletion coexisting with FTO and MC1R gene variants, causes severe early onset obesity. Hormones (Athens) 2016, 15(3):445-452.
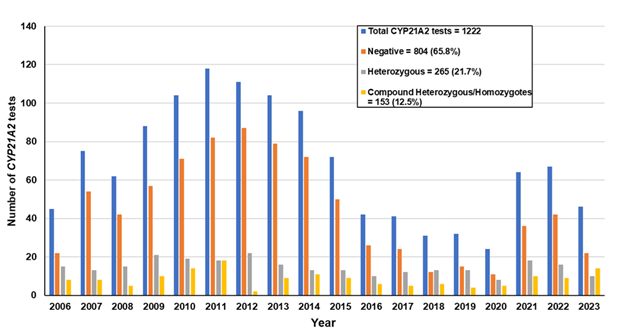
Εικόνα 1. Συνολικά 1222 ασθενείς με κλινική υποψία CAH έχουν εξεταστεί από το 2006 έως τον Ιούλιο του 2023 για μεταλλάξεις στο γονίδιο CYP21A2, διακόσοι εξήντα πέντε (21.7%) αναγνωρίστηκαν ως ετεροζυγώτες και 153 (12.5%) ως σύνθετοι ετερωζυγώτες/ομοζυγώτες.
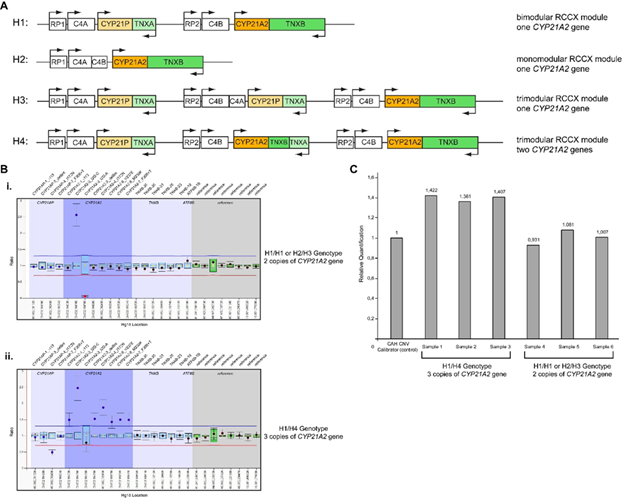
Εικόνα 2.
(Α) Σχηματική αναπαράσταση των τεσσάρων διαφορετικών απλοτύπων που προσδιορίστηκαν στην μελέτη του ΜΓΛΘ [17]. H1: Απλότυπος 1, διμερής μονάδα RCCX με ένα γονίδιο CYP21A2; H2: Απλότυπος 2, μονομορφική μονάδα RCCX με ένα γονίδιο CYP21A2; H3: Απλότυπος 3, τριμερής μονάδα RCCX με ένα γονίδιο CYP21A2; H4: Απλότυπος 4, τριμερής μονάδα RCCX με δύο γονίδια CYP21A2.
(Β) MLPA διαγράμματα αναλογιών από ασθενείς που προσδιορίζονται ως φορείς για την παθογόνο παραλλαγή p.Gln319Ter που δείχνουν τους γονότυπους i) H1/H1 ή H2/H3 και ii) H1/H4 στο χρωμόσωμα 6p21.3.
(C) CAH Real Fast CNV Assay ασθενών που προσδιορίστηκαν ως φορείς για την παθογόνο παραλλαγή p.Gln319Ter που δείχνει τον γονότυπο H1/H4 (δείγματα 1-3) και τον γονότυπο Η1/Η1 ή Η2/Η3 (δείγματα 4-6).
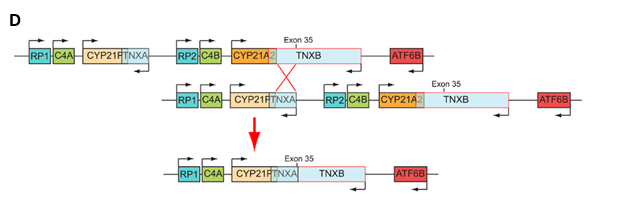
(D) Σχηματική αναπαράσταση του συμπλέγματος RCCX στο χρωμόσωμα 6p21.3 στο οποίο τα γονίδια TNXA και TNXB υφίστανται άνισο επιχιασμό με αποτέλεσμα τη διμιουργία χίμαιρας τύπου TNXA/TNXB CH-1 σε Κύπριους ασθενείς με την κλασική μορφή CAH με απώλεια άλατος (Salt Wasting-SW).
Εικόνα 3. Επιπολασμός των διαφορετικών παραλλαγών του CYP21A2 στον κυπριακό πληθυσμό. Εντοπίστηκαν συνολικά 571 παθογόνες παραλλαγές, με την παραλλαγή p.Val281Leu να είναι η πιο συχνή (62,17%) στους ασθενείς που μελετήθηκαν (n=418).
Πίνακας 1. Λεπτομερής ανάλυση συσχέτισης γονότυπου/φαινοτύπου σε ασθενείς με την κλασική απώλεια άλατος (SW) και την απλή αρρενωποιητική (SV) μορφή CAH. Όλοι οι ασθενείς υποβλήθηκαν σε γενετικό έλεγχο στο γονίδιο CYP21A2.
Πίνακας 2. Κλινικά και γενετικά χαρακτηριστικά των ελεγχθέντων ασθενών με ΣΥΥ με επτά από αυτούς να φέρουν νέες μεταλλάξεις
Εικόνα 4. Α. Από 517 ασθενείς με ύποπτο MEN2 (2002-σήμερα), το 11.2% (58 άτομα) είχαν παθογόνο παραλλαγή στο πρωτο-ογκογονίδιο RET. Β. Η ανάλυση απλότυπου με μικροφορικούς δείκτες έδειξε κοινό απλότυπο σε 12 οικογένειες με p.Cys618Arg, πιθανό αποτέλεσμα του φαινομένου της αρχής του ιδρυτού (founder effect) στην Κύπρο.
Εικόνα 5. Η φυσιολογική εφηβεία ξεκινά στις ηλικίες 8-13 στα κορίτσια και 9-14 στα αγόρια. Σχετίζεται με σωματικές αλλαγές, όπως η ανάπτυξη του στήθους (θηλαρχή) και η έναρξη της εμμήνου ρύσεως (εμμηναρχή) στα κορίτσια, καθώς και αλλαγές στη φωνή και μεγέθυνση των όρχεων στα αγόρια. Και στα δύο φύλα παρατηρείται ανάπτυξη των γεννητικών οργάνων, και εμφάνιση τριχοφυΐας στο εφήβαιο και στις μασχάλες.
Εικόνα 6. Α) Σχηματική αναπαράσταση της φυσιολογικής γενωμικής αποτύπωσης (genomic imprinting) του γονιδίου MKRN3: σίγηση του μητρικού αλληλόμορφου μέσω μεθυλίωσης στις CpG νησίδες του υποκινητή (promoter) και έκφραση του πατρικού αλληλόμορφου. (Β) Αναπαράσταση πατρικής κληρονομικής μετάλλαξης με χαρακτηριστική απώλεια λειτουργίας (paternal inherited loss-of-function mutation). Και τα δύο αλληλόμορφα είναι ανενεργά αφού το πατρικό μεταλλάσσεται και το μητρικό αποσιωπάται.
Εικόνα 7. Σχηματική αναπαράσταση των μεταλλάξεων που εντοπίστηκαν στο γονίδιο MKRN3 από το εργαστήριο ΜΓΛΘ του ΙΝΓΚ.






0002.jpg)